Series Editor: Mike O’Brien, PharmD
Quick evidence-based pearls on all things pharmacology and pharmacy-related in Emergency Medicine

Series Editor: Mike O’Brien, PharmD
Quick evidence-based pearls on all things pharmacology and pharmacy-related in Emergency Medicine

Two new oral agents were given Emergency Use Authorization to be used in patients with mild-moderate COVID-19 at high risk of progression to severe infection, molnupiravir and nirmatrelvir/ritonavir (Paxlovid) [1,2]. Prior to this authorization, most evidence-based COVID therapies were parenteral and required significant healthcare resources to coordinate and administer.
| Nirmatrelvir/ritonavir [3] | Molnupiravir [4] | |
|---|---|---|
| Mechanism |
Protease inhibitor leadings to interruption of viral replication Ritonavir has no role in treating COVID-19, it is only included to boost levels of nirmatrelvir via CYP3A4 inhibition |
Increased frequency of RNA mutations and impaired replication [5] |
| Efficacy vs Placebo (Hospitalization or Death) | 0.8% vs 6.3% (CI -7.21 to -4.03) | 6.8% vs 9.7% (CI -5.9 to -0.1) |
| Drug Interactions | CYP3A4 inducers, inhibitors, and substrates
May decrease efficacy of hormonal contraceptives, non-hormonal contraceptives should be considered Contraindicated medications include: amiodarone, carbamazepine, clozapine, colchicine, dihydroergotamine, dronedarone, flecainide, lovastatin, ranolazine, sildenafil, simvastatin Many other important interactions exist so care should be taken to assess all medication interactions |
N/A |
| Cost* | Patient: $0
US government: $530 [6] |
Patient: $0 US Government: $700 [7] |
| Dose | 300 mg/100 mg BID for 5 days
Must be started within 5 days of symptom onset |
800 mg BID for 5 days
Must be started within 5 days of symptom onset |
| Notes | Approved for patients ≥ 12 years old AND ≥ 40 kg
Not approved for inpatient initiation If patient is hospitalized, continuation is up to the discretion of the provider Not used as pre-/post-exposure prophylaxis |
Approved for patients ≥ 18 years
Not approved for inpatient initiation If patient is hospitalized, continuation is up to the discretion of the provider Not used as pre-/post-exposure prophylaxis |
| Renal/Hepatic Dose Adjustments | eGFR ≥30 to <60 mL/min: 150 mg/100 mg BID
eGFR <30 mL/min: Not recommended Child-Pugh class C: Not recommended |
None |
*Note: The US federal government has purchased 10 million doses of nirmatrelvir/ritonavir and 3 million doses of molnupiravir [8,9]. These supplies will be allocated to states and territories as needed and will be available to patients at no charge.
Nirmatrelvir/ritonavir (Paxlovid)
Paxlovid was evaluated in the EPIC-HR trial, which is not fully published at this time [3]. This was a phase 2/3, double-blinded, randomized placebo controlled trial including nonhospitalized, unvaccinated patients adults with mild-moderate COVID-19 within 5 days of symptom onset with at least 1 risk factor† for development of severe illness from COVID-19. Exclusion criteria included patients with a history of COVID-19 infection or COVID vaccination. Patients were given Paxlovid 300 mg/100 mg or placebo BID for 5 days. The primary outcome was hospitalization or death at day 28. The modified intention-to-treat1 (mITT1) group excluded patients who did not receive nor were expected to receive COVID-19 mAb treatment. In the mITT1 group, the primary outcome occurred in 0.8% of patients receiving Paxlovid vs 6.3% of patients in the placebo group (8/1039 vs 66/1046, CI -7.21 to -4.03).
These results appear quite robust with a fragility index of 37. Additionally, in patients with detectable COVID antibodies there was less of an impact of the study medication. However, these patients still appeared to have some benefit (0.2% vs 1.5%, CI -2.45 to -0.23) which suggests that vaccinated patients may still benefit from Paxlovid.
†Risk factors for progression to severe disease: BMI >25, chronic lung disease, asthma, chronic kidney disease, current smoker, immunosuppressive disease or immunosuppressive treatment, cardiovascular disease, hypertension, sickle cell disease, neurodevelopmental disorders, active cancer, medically-related technological dependence, or age >60 years
Molnupiravir
Molnupiravir was evaluated in the MOVe-OUT trial [10]. This was a phase 3, double-blinded, randomized, placebo controlled trial including nonhospitalized, unvaccinated adults with mild-moderate COVID-19 within 5 days of symptom onset with at least 1 risk factor‡ for development of severe illness from COVID-19. Exclusion criteria included anticipated hospitalization within 48 hours, eGFR <30 or dialysis dependent, pregnancy, and COVID vaccination. Patients were able to receive steroids but not monoclonal antibodies (mAbs) nor remdesivir. Patients were given molnupiravir 800 mg or placebo BID for 5 days. The primary outcome was hospitalization or death at 29 days. In the mITT population, the primary outcome occurred in 6.8% of patients in the study group vs 9.7% in the placebo group (48/709 vs 68/699, CI -5.9 to -0.1). Death occurred in 1 patient on molnupiravir and in 9 patients on placebo (0.1% vs 1.3%, RRR 89%, CI 14 to 99).
Despite the above results, this may not be the positive trial it initially appears. First of all, for the primary outcome, the fragility index is 0, meaning that if 1 more patient in the study group experienced the primary outcome then it would have changed the statistical significance. Additionally, when the mITT analysis was adjusted for sex, the absolute risk reduction remained 2.8% but the confidence interval was not significant (-5.7 to 0.1). Lastly, in the subgroup analysis, there was no benefit in patients that had positive COVID antibody tests and there was a slight preference towards placebo over molnupiravir (3.7% vs 1.4%, ARR 2.3, CI -1.7 to 7.1). This suggests that vaccinated patients may not benefit from this therapy as much (or at all) as compared to unvaccinated patients.
‡Risk factors for progression to severe disease: age >60 years, active cancer, chronic kidney disease, COPD, BMI ≥30, heart failure, coronary artery disease, cardiomyopathy, or diabetes mellitus
Note: Both the EPIC-HR and MOVe-OUT studies were funded by their respective pharmaceutical company.
Read other articles in the EM Pharm Pearls Series and find previous pearls on the PharmERToxguy site.

Many guidelines and treatment algorithms for diabetic ketoacidosis (DKA) recommend sodium chloride 0.9% as the replacement fluid of choice, though alternative fluids may be a better option [1-4]. Randomized trials, in adult and pediatric patients, demonstrate faster resolution of DKA when using balanced solutions (e.g., PlasmaLyte-A, lactated Ringer’s) compared to sodium chloride [5-7]. Dr. Josh Farkas provides further review of this topic in 3 excellent and detailed EMCrit posts [8-10].
A phase-2 study published in 2021, SCOPE-DKA, randomized 93 patients with severe DKA (median venous pH 7.0) to receive PlasmaLyte-148 (PlasmaLyte-A) or sodium chloride 0.9% [11]. During the first 48 hours of treatment, patients received a average of ~6.5 L of fluid. At 24-hours, more patients in the PlasmaLyte group had resolution of DKA (defined as base excess ≥ -3 mEq/L) as compared to the sodium chloride group (69% vs 36%, p=0.002). However, by 48-hours, both groups had similar rates of DKA resolution (96% vs 86%, p=0.111). The study authors concluded that PlasmaLyte-148 may lead to faster resolution of metabolic acidosis in patients with DKA without an increase in ketosis, in line with findings from previous studies, but these results need to be confirmed in a larger, Phase 3 trial.
To further explore the nuances, strengths, and weaknesses of this study, please read the REBEL EM review by Dr. Mark Ramzy [13].
Read other articles in the EM Pharm Pearls Series and find previous pearls on the PharmERToxguy site.

Early antibiotics are recommended for treatment of many infections, including patients with sepsis or septic shock [1]. Critically-ill patients and those with a suspected infection at risk for severe illness are generally administered two (or more) empiric antibiotics in the emergency department (ED) which cover a wide range of potential pathogens. A typical approach includes utilizing a broad-spectrum antibiotic (frequently a beta-lactam such as cefepime or piperacillin-tazobactam) plus an anti-MRSA agent (typically vancomycin).
Early in the patient’s hospital stay they may have limited IV access, so the question often arises as to which antibiotic to give first, the broad-spectrum antimicrobial or the anti-MRSA agent. Additionally, though the overall risk of an allergic reaction is relatively low with most antimicrobials, when multiple agents are given simultaneously it can be difficult to ascertain which one may have caused a reaction and lead to incorrectly documented allergies, so it can be important to consider if the initial doses should be administered separately. However, there isn’t strong data to guide practice in terms of giving the initial antibiotics concurrently vs consecutively, from an allergy perspective. To further complicate the issue, patients may also develop delayed reactions so a strong causal relationship cannot always be determined. In practice, there are times (increasingly so with rising ED patient volumes) when we give antibiotics one at a time simply for logistical reasons. So that begs the question, which antibiotic should be given first?
In patients with sepsis or septic shock, early antibiotics significantly decrease mortality [1]. This relationship is strongest for patients with septic shock, where the odds of in-hospital mortality was increased by 1.04-1.16 for each hour antibiotics were delayed [2-4]. Notably, broad spectrum antibiotics are deemed such as they cover both gram positive and gram negative pathogens, therefore the addition of an anti-MRSA agent contributes a relatively smaller amount of coverage and is primarily targeted at resistant gram-positive bacteria. Additionally, gram-negative pathogens tend to cause a higher degree of illness and mortality, so it would be reasonable to give the broad-spectrum antibiotic first [5-7]. As both cefepime and piperacillin-tazobactam are recommended to be infused over 30 minutes (though this can vary based on institutional policies) and vancomycin is typically infused over 1 hour for each gram, if vancomycin is administered first, patients may wait hours to receive a broad spectrum agent.
A recent study now supports this practice. This is an observational trial which evaluated 3,376 patients with a blood stream infection, 2,685 patients received a beta-lactam first and 691 patients received vancomycin first [8]. They found that patients who received a beta-lactam prior to vancomycin had significantly improved 48-hour and 7-day mortality. Further review of this article may be found on the JournalFeed blog post.
Bonus tip: Having antibiotics stocked on the unit reduces time to administration [9].
Read other articles in the EM Pharm Pearls Series and find previous pearls on the PharmERToxguy site.

Treatment of digoxin toxicity can be quite complex and generally involves the use of digoxin immune Fab (DigiFab®) for symptomatic patients. The dosing of DigiFab can vary depending on the amount ingested, serum concentration, and/or suspected chronicity of toxicity. Alternatively, for an acute ingested of an unknown amount where the serum concentration is not available, it is recommended that 10 vials of DigiFab be administered empirically. This antidote is expensive (~$5,000 per vial) and not always readily available in every hospital. Given the complicated dosing and cost, alternative dosing strategies are being explored.
Researchers from Australia first proposed an initial 2-vial DigiFab dose for acute digoxin poisoning in a 2014 review article [1]. They followed this up with a pharmacokinetic study supporting the simplified dosing scheme [2]. Based on their early data, the Australian poison center recommendations were revised to instead use small doses of DigiFab (2 vials at a time) with repeat doses as needed to achieve clinical effect. This allowed them to prospectively study this new dosing strategy in 21 cases of digoxin toxicity [3]. Most patients required less than would have been administered following traditional dosing calculations. Patients receiving the lower-dosing scheme did have a rebound in free digoxin levels >2 ng/mL at a median time of 18 hours in patients with normal renal function and 103 hours in patients with an acute kidney injury. Most patients received 2 vials of DigiFab initially and a median of 4 vials total after receiving additional doses based on persistent or recurrent symptoms. Overall, patients required significantly less antidote with similar clinical outcomes. Importantly, there are limitations with the data to date, highlighted in a letter-to-the-editor with a subsequent response from the original authors [4, 5]. This titration approach should only be considered with input from a toxicologist and still requires the same level of monitoring.
| Characteristics and Savings | |
|---|---|
| Amount of digoxin ingested* | 13 mg (9.5-25 mg) |
| Initial potassium* | 5 mEq/L (4.5-5.4 mEq/L) |
| Fatalities due to digoxin toxicity | 0 |
| Estimated vials saved^ | 223-356 vials |
| Estimated cost savings^† | $1.1-1.8 million |
* Median (IQR)
|
|
Following administration of DigiFab, avoid measurement of the total digoxin concentration as this measures both free drug and drug bound to DigiFab, which will cause the result to be falsely elevated [6]. Additionally, extracorporeal treatments are not recommended for the removal of digoxin or the digoxin-Fab complex, regardless of the clinical context [7].
Read other articles in the EM Pharm Pearls Series and find previous pearls on the PharmERToxguy site.

Sodium bicarbonate during a cardiac arrest is widely debated and used in many cases. In a 2018 PULMCrit post, Dr. Josh Farkas reviews much of the data and concludes that use of sodium bicarbonate is a “source of eternal disagreement.” A 2013 EMCrit article and podcast by Dr. Scott Weingart also details some of the controversy. The 2020 ACLS Guidelines state that routine use of sodium bicarbonate is not recommended in cardiac arrest [1]. Despite this recommendation, sodium bicarbonate is still often administered during resuscitations if a metabolic (or respiratory) acidosis is suspected or after a prolonged downtime. A recent study evaluated the effect of pre-arrest acid-base status on response to sodium bicarbonate and achievement of return of spontaneous circulation (ROSC) [2].
This was a retrospective review of in-hospital cardiac arrests (IHCA) in patients with pre-arrest serum bicarbonate levels ≤21 mmol/L compared to >21 mmol/L. Pre-arrest bicarbonate levels were obtained <24 hours prior to the arrest. Similarly, post-arrest bicarb levels were obtained <24 hours following the arrest. Bicarbonate levels were recorded from basic chemistry panels rather than blood gases. All patients received a median sodium bicarbonate dose of 100 mEq. The groups were relatively well-matched, with the only major difference being the time to first bicarb administration was faster in the ‘acidotic’ group (6.9 vs. 9.2 minutes). Initial ECG rhythms were similar between the groups.
Read other articles in the EM Pharm Pearls Series and find previous pearls on the PharmERToxguy site.

Bleeding patients or those undergoing procedures that are at high risk of bleeding may require correction of their INR. Multiple products can be used to achieve this, including fresh frozen plasma (FFP). FFP contains many substances, including clotting factors, fibrinogen, plasma proteins, electrolytes, and anticoagulant factors. It is sometimes said that the intrinsic INR of FFP is approximately 1.6-1.7 and that it’s not possible to achieve a lower INR. This pearl will further explore these concerns.
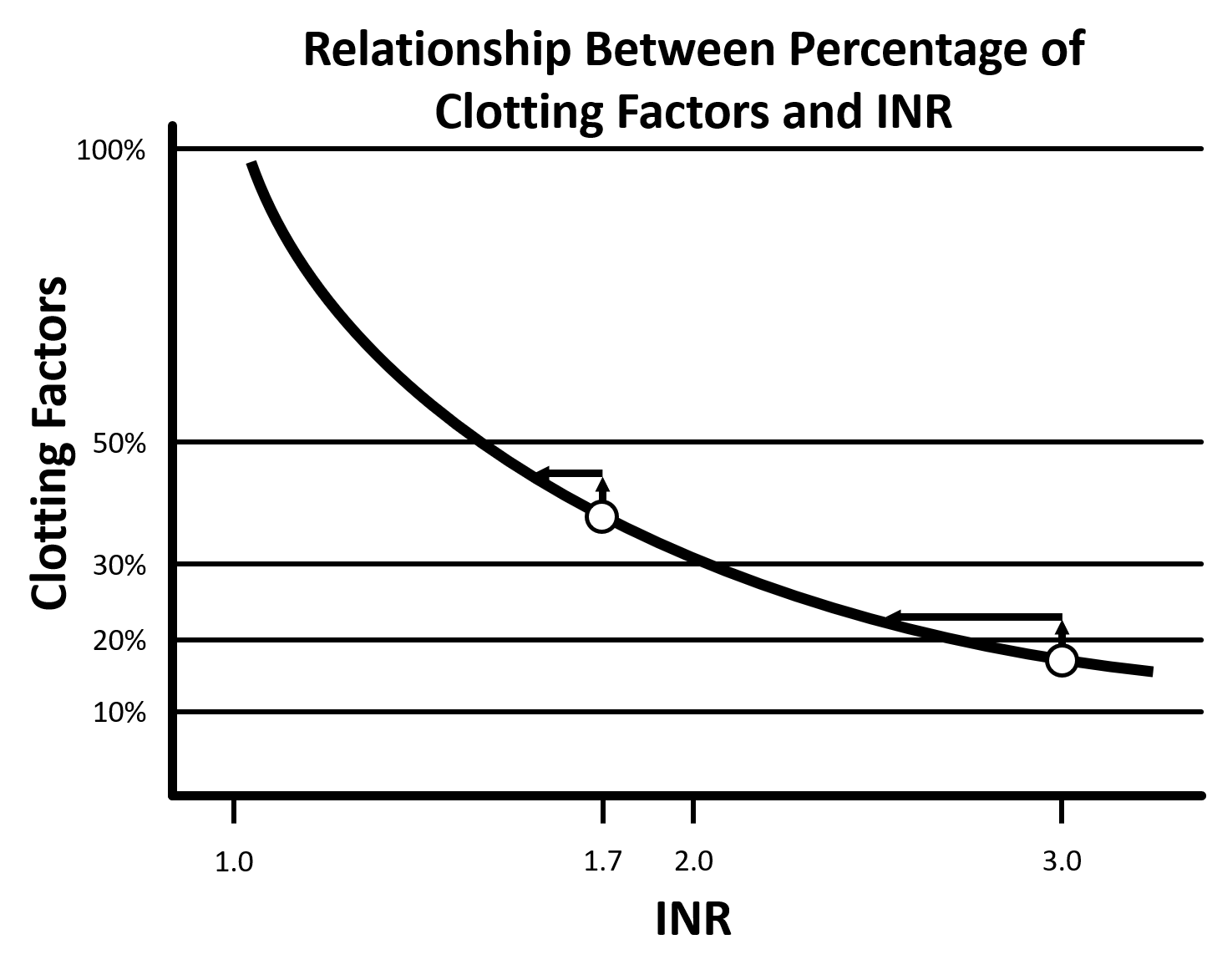
Figure 1: Adapted from Dzik 2012 [7].
| Amount of FFP to Achieve a Target INR Based on Pre-FFP INR | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Target INR | |||||||||
| 1.3 | 1.7 | 3.0 | |||||||
| Initial INR | Volume (L) | Dose (mL/kg) | Factor (%) | Volume (L) | Dose (mL/kg) | Factor (%) | Volume (L) | Dose (mL/kg) | Factor (%) |
| 6.0 | 4.5 | 64 | 45 | 2.5 | 36 | 25 | 1.5 | 21 | 15 |
| 5.0 | 4.3 | 61 | 43 | 2.3 | 32 | 23 | 1.0 | 14 | 10 |
| 4.0 | 4.0 | 57 | 40 | 2.0 | 29 | 20 | 0.5 | 7 | 5 |
| 3.0 | 3.5 | 50 | 35 | 1.5 | 21 | 15 | – | – | – |
| 2.0 | 2.5 | 36 | 25 | 0.5 | 7 | 5 | – | – | – |
Table 1: Adapted from Holland 2006 [3]. Note: 1 unit of FFP is ~200-250 mL
Read other articles in the EM Pharm Pearls Series and find previous pearls on the PharmERToxguy site.

Succinylcholine is frequently used in the ED to facilitate intubation, but it may be avoided in some cases due to the risk of hyperkalemia. The underlying physiology of this effect appears to be directly related to its therapeutic mechanism of action. When succinylcholine binds to and activates acetylcholine receptors, it leads to an influx of sodium and calcium and and an efflux of potassium into the extracellular space [1]. Additionally, when these acetylcholine receptors are immature or denervated, it seems that these channels may stay open significantly longer, allowing for an increased amount of potassium to exit the cell, leading to an increased risk of hyperkalemia.
Based on multiple studies that included patients with normal renal function, succinylcholine leads to a serum potassium increase of ~0.5 mEq/L [2-4]. This is likely clinically insignificant in most patients. In fact, an ED-based study found a variable response with serum potassium increasing in 46 cases, decreasing in 46 cases, and not changing in 8 cases [3]. It seems that even patients on chronic dialysis are not at increased risk of developing clinically-significant hyperkalemia from succinylcholine [5].
So, when should succinylcholine potentially be avoided specifically due to hyperkalemia concerns [6]?
In patients for whom succinylcholine is determined to be not an option, non-depolarizing muscular blocking agents (NMBAs), such as rocuronium, are still safe and do not lead to hyperkalemia.
Read other articles in the EM Pharm Pearls Series and find previous pearls on the PharmERToxguy site.